Hydrogenation of Graphene
Graphene is presently the hottest material in the world. It is a perfect mesh of carbon six-rings, in which all the C-atoms are sp2 hybridised. It has amazing properties: large electron mobility, strong mechanical stability, .... Because of that Geim and Novoselov, who were the first to find a way to make graphene available for studies in the laboratory, were honoured with the Nobel prize in 2010.
Electronically, graphene is a semi-conductor, albeit with a 0 Volt band gap. That is an interesting property for physicists, but not that useful if one wants to build a transistor from graphene. In order to switch a current, one needs a finite band gap. One way to achieve this is to hydrogenize graphene in a regularly patterned fashion. With this motivation in mind we studied hydrogenation of graphene.
Only hydrogen atoms as radicals are able to form bonds with graphene, not molecules. And even this reaction is slow, as the C-atom has to rehybridize from sp2 to sp3. Following the general rules about binding to ring-conjugated hydrocarbons, it is expected that the 2nd hydrogen atom binds in ortho- or paraposition, whereas the meta-position is energetically unfavourable. The binding energies of H in these geometries is much smaller than in a typical hydrocarbon, because there is a large stress in the structure.
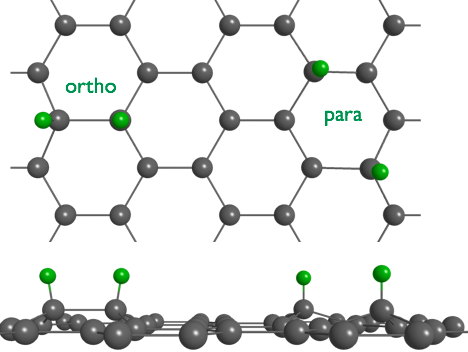
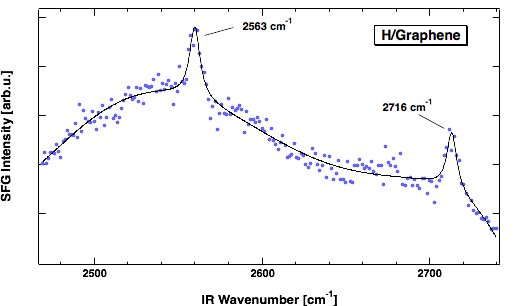
We use sumfrequency spectroscopy (SFS) to obtain a vibrational spectrum from a molecular sheet such as graphene. SFS is a technique in laser spectroscopy which selectively detects with high sensitivity bonds at surfaces. In the spectrum we find two lines separated by 153 cm-1. One at 2563 and one at 2716 cm-1. The first corresponds to para- and the 2nd to ortho-dimers of hydrogen. The observed line positions are in excellent agreement with calculations by S. Sakong and P. Kratzer, Physics department, allowing us an unambiguous assignment. The rather low wavenumbers, when compared to the typical 3000 cm-1 for C-H bonds, are inline with the small binding energy of H in this structure.
But is not the end of the story. In fact, it is way more interesting. We are not able to study a sheet of graphene suspended in vacuum. Rather we adopted a technique where on prepares a graphene by pyrolysis of C2H4 on Ir. More precisely, we use an Ir crystal with the hexagonal, (111), surface exposed to vacuum. On this surface the unit cell is about 10% larger than the mesh size of graphene. Hence, a Moire pattern results. In this superstructure one finds areas where every 2nd C-atoms is located above an Ir-atom, and such where the center of a C-ring falls on top of an Ir atom.
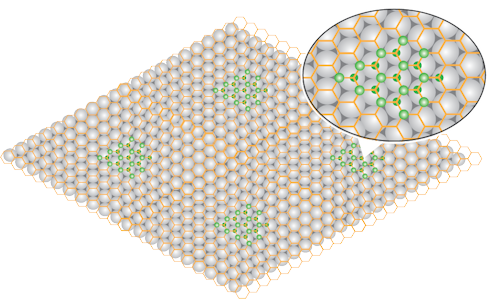
What we find is that H preferentially binds in the first of these two areas, and binds from both sides of the graphene sheet. The resulting structure corresponds to a saturated hydrocarbon and is therefore called
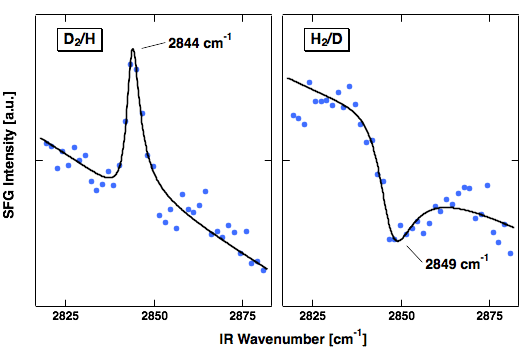
Moreover, at the Ir sites open to vacuum not only H atoms adsorb but also H2 molecules are dissociatively adsorbed. This allowed for an intriguing experiment. We dosed first molecular hydrogen in order to fill the space between the Ir surface and graphene sheet with atoms. Then we exposed the system to the other isotope of hydrogen. This allows us to offer H from the bottom and D from the vacuum side or vice versa. Hence, we can prepare graphene with graphane areas of selected isotopes bound to the two sides.
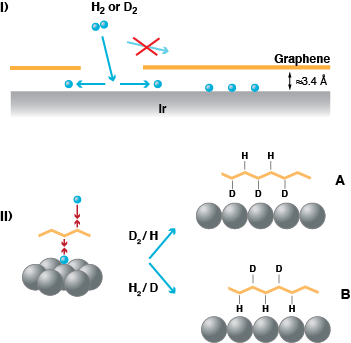
SF spectroscopy allows us to experimentally prove that this is the case. As it is a coherent technique the vibrational line appears as peak or as a dip on the background signal depending on the orientation of the bond which is vibrationally excited. We observe a peak when we dose D2 first and then later on H, we find a dip when we dose H2 and then D atoms. The vibrational line is found at 2844 cm-1 for H, a value close to what is known for H bound to diamond. Hence, it is indicative of binding to a completely sp3-rehybridised C-atom as one would expect for graphane.
Our results are consistent with studies carried out by Liv Hornekær in Århus using scanning probe microscopy. In the micrographs obtained one sees that the bound H atom cluster in specific areas of the Moire pattern. They are able to identify that these are the areas where every 2nd C atoms sits above an Ir atom.

STM micrograph of a hydrogenated graphene sheet on Ir(111). The bright areas are where H is bound. Note the lage image scale! What you are seeing is not the hexagonal structure of graphene, but the one of the Moire pattern. (Balog et al., Nature Mat. 9, 315 (2010))
The graphane areas are what dominates hydrogen adsorption to epitaxial graphene on Ir(111). The dimers we observed first are minority species. However, the hydrogen in the graphene areas escape detection in SFS, when a single hydrogen isotope is used, as the detection is forbidden due to the centro-symmetry, when H is bound on both sides of the graphene sheet.
If you want to learn more, have a look at our recent publications.
